L'ophtalmologie et la génétique sont deux disciplines qui ne cessent de s'enrichir l'une l'autre. L'histoire de la génétique ophtalmologique le démontre de la fin du XIXesiècle, avec la description princeps de maladies rétiniennes familiales et de leurs associations syndromiques, à la découverte de la double hélice d'ADN en 1953 en passant par la découverte de nombreux gènes impliqués dans des maladies oculaires en fin de XXesiècle. Aujourd'hui, ce sont les succès époustouflants de la biologie fondamentale et appliquée aux thérapies et renforcés par les progrès de l'intelligence artificielle qui explosent en ce premier quart de XXIesiècle.
La Société Française d'Ophtalmologie ne s'est pas trompée en sollicitant régulièrement des rapports sur les maladies génétiques ophtalmologiques qui sont un parfait miroir d'avancées médico-scientifiques majeures.
En 1963, Albert Franceschetti, Jules François et Jean Babel présentent leur ouvrage Les Hérédo-dégénérescences chorio-rétiniennes (Dégénérescences tapéto-rétiniennes) . Ce rapport brillait de mille feux d'intelligence clinique ophtalmologique et génétique alors que la biologie moléculaire était naissante.
En 2005, le rapport de Jean-Louis Dufier et Josseline Kaplan Œil et génétique illuminait les importantes découvertes scientifiques portées par la révolution des outils de biologie moléculaire, avec au premier chef les découvertes de gènes impliqués dans les maladies ophtalmiques héréditaires et les conséquences immédiates pour le conseil génétique.
En 2026, quelque vingt années plus tard, un arrêt sur image s'impose, à nouveau, sur le domaine des dystrophies génétiques de la rétine et du nerf optique, qui incarnent parfaitement la symbiose de l'accélération des connaissances scientifiques et médicales destinées in fine à chaque personne malade.
En 20 ans, le paysage pour les maladies rares en France s'est métamorphosé avec des plans nationaux successifs (Plans Nationaux Maladies Rares) et la création de centres de référence (CRMR) notamment dédiés aux maladies rares sensorielles dans le cadre de la Filière de Santé Maladies Rares SENSGENE, construite grâce à l'expertise de cliniciens hors pair qui ont colligé, dans ce rapport, leurs savoirs des quatre coins de la France à partir d'équipes cliniques et de laboratoires de diagnostic génétique qui assurent avec brio le parcours de soins de ces malades sur le territoire.
De plus, au cours des toutes dernières années, la France s'est dotée d'un Plan France Médecine Génomique (PFMG) qui permet l'accès au séquençage du génome à visée diagnostique constituant un pas de géant pour la connaissance des causes génétiques des maladies. Le domaine des dystrophies rétiniennes en bénéficie tout particulièrement en raison du grand nombre de gènes impliqués ainsi que de la complexité des corrélations entre les aspects cliniques et les causes génétiques.
Tous ces progrès sont le résultat de travaux scientifiques d'équipes françaises et internationales pour découvrir les gènes qui, mutés, entraînent ces maladies, pour comprendre les mécanismes physiopathologiques responsables au niveau des cellules rétiniennes grâce à une multitude de modèles in vitro ou in vivo et, enfin et surtout, pour concevoir des stratégies thérapeutiques. Toutes ces avancées n'ont qu'un but final, celui de pouvoir traiter ou, en tout cas, ralentir les processus en cause dans les dégénérescences rétiniennes ou du nerf optique.
En 20 ans, les développements de stratégies thérapeutiques ont été fulgurants. Le succès de la thérapie génique de remplacement pour une forme de dystrophie rétinienne très précoce liée au gène RPE65 grâce au Luxturna® (voretigene neparvovec) est démontré et accessible aux patients désormais dans de nombreux pays (en France dès 2019). Ce résultat pionnier a ouvert la voie aux thérapies tant attendues. De nouveaux traitements sont fondés, en plus de la thérapie génique classique où un vecteur viral délivre le gène, sur des approches innovantes très variées, allant du ciblage précis d'une mutation avec des « velcros » moléculaires (oligonucléotides anti-sens) ou à l'aide de « ciseaux » et de système de réparations (CRISPR-Cas9) à l'activation artificielle de cellules rétiniennes épargnées (avec l'optogénétique). Malgré ces progrès précliniques extraordinaires, le chemin vers le malade reste long et complexe, surtout si l'on arrive au stade des essais cliniques dont ledesign doit prendre en compte la grande hétérogénéité de ces maladies pour lesquelles un ralentissement ou un arrêt du processus dégénératif doit être prouvé aux autorités réglementaires, ce qui peut représenter un véritable défi.
Nous remercions la Société Française d'Ophtalmologie de nous permettre ce coup de projecteur sur les maladies héréditaires de la rétine et du nerf optique pour lesquelles les approches thérapeutiques sont devenues des réalités cliniques, notamment au vu des nombreux essais cliniques en cours.
Ce rapport est riche de contributeurs d'horizons très variés témoignant de l'importance de l'approche holistique dont doivent, toujours et plus que jamais, pouvoir bénéficier les malades : des ophtalmologistes experts du domaine, des scientifiques du plus haut niveau et aussi des spécialistes de rééducation et de prise en charge psycho-sociale. En effet, un diagnostic de dystrophie rétinienne génétique a des répercussions personnelles et familiales qui dépassent l'aspect purement médical ; le conseil génétique, le soutien psychologique, la rééducation restent incontournables.
Acteurs quotidiens de la prise en charge des malades et experts en pathologies du segment postérieur, nous remercions très vivement, ainsi que leurs équipes (voir la liste détaillée des auteurs et collaborateurs), les très nombreuses personnes qui ont construit avec nous ce rapport et qui œuvrent au sein des hôpitaux français : CHU de Lille (Claire-Marie Dhaenens, Vasily Smirnov, Sabine Defoort-Dhellemmes, Isabelle Drumare), CHU de Montpellier (Isabelle Meunier, Anne-Françoise Roux, Béatrice Bocquet), CHU d'Angers (Patrizia Amati-Bonneau), CHU de Bordeaux (Benoît Arveiler, Vincent Michaud), CHU de Nantes (Guylène Le Meur), CHU de Rennes (Xavier Zanlonghi), l'Assistance Publique-Hôpitaux de Marseille (AP-HM), Hôpital de la Timone (Aline Cano), le CHU de Strasbourg (Thomas Wirth, Marie-Thérèse Abi Warde, Vincent Laugel, Mathieu Anheim, Chloé Trouvé-Laeng, Valérie Pelletier) ; et, bien entendu, les hôpitaux parisiens, avec l'Assistance Publique-Hôpitaux de Paris (AP-HP) : le département de la recherche clinique et du développement (Isabelle Durand-Zaleski), l'Hôpital Necker-Enfants Malades (Alejandra Daruich, Mathieu Robert, Dominique Bremond-Gignac, Sophie Valleix, Sandrine Marlin), l'Hôpital Cochin (Francine Behar-Cohen), l'Hôpital Beaujon (Nadia Belmatoug) ; avec la Fondation Rothschild (Catherine Vignal-Clermont, Élise Boulanger-Scemama, Elsa Laumonier Demory, Yannick Le Mer, Dan Miléa), avec l'Hôpital intercommunal de Créteil (Alexandra Miere, Éric Souied) et, enfin, l'Hôpital des 15-20 (Anne-Élisabeth Chaumet-Riffaud, Philippe Chaumet-Riffaud, Pierre-Olivier Barale, Béatrice Le Bail, Christophe Pourchez, Anaëlle Carou, Camille Andrieu). Ce rapport a été enrichi par la contribution d'experts cliniciens internationaux francophones, Elise Heon (Sick Kids Hospital, Toronto, Canada), Bart P. Leroy (Hôpital Universitaire de Gand, Belgique) et Elias I. Traboulsi (Cleveland Clinic, Cleveland, États-Unis).
Les équipes de recherche françaises sont bien au rendez-vous face aux défis scientifiques de ces maladies et elles ont couvert tous les aspects de la traque des gènes non identifiés, en passant par la dissection des dysfonctions jusqu'aux approches thérapeutiques. Nous saluons ainsi la remarquable contribution de chercheurs français et internationaux, très notamment : Jean Bennett (Université de Pennsylvanie, États-Unis), Kinga Bujakowska (Harvard Medical School, États-Unis), Pascal Escher (Université de Bern, Suisse) ; et pour la France : Guy Lenaers (Université d'Angers), Jean-Michel Rozet, Isabelle Perrault (Institut IHU IMAGINE, Paris), Vasiliki Kalatzis (Institut des neurosciences de Montpellier, Montpellier), Camille Beluffi-Marin (Institut de Génétique Médicale d'Alsace, Strasbourg), Aziz El-Amraoui (Institut Pasteur, Paris), Christelle Monville (Université Paris Saclay), et enfin de l'Institut de la Vision avec Christina Zeitz, Olivier Goureau, Colas Authié, Deniz Dalkara, Filippo Del Bene, Xavier Guillonneau, Alexandra Rebsam et Serge Picaud.
Nos remerciements vont aussi tout particulièrement vers les précieuses aides organisationnelles : avec Larissa Moutsimilli (chargée de mission SENSGENE, Hôpital des 15-20, Paris) et le pilotage de Marilyne Oswald (cheffe de projet SENSGENE, CHU de Strasbourg).
Ce rapport, Des gènes aux traitements , est un nouveau point d'étape dans un domaine bouillonnant d'innovations, et dont les perspectives d'applications pour les malades vont crescendo et nous obligent avant tout à comprendre leurs besoins. Les mots bouleversants de Christina Fasser (présidente sortante de Retina International), une patiente experte des maladies rétiniennes, nous rappellent l'importance de prendre en compte chaque malade, chaque histoire, chaque personne…
La médecine génétique personnalisée en ophtalmologie est en train de naître. L'histoire ne fait que commencer…
Nous vous souhaitons une bonne lecture et dédions ce rapport à nos malades et leurs familles.
Hélène Dollfus
Isabelle Audo
José-Alain Sahel
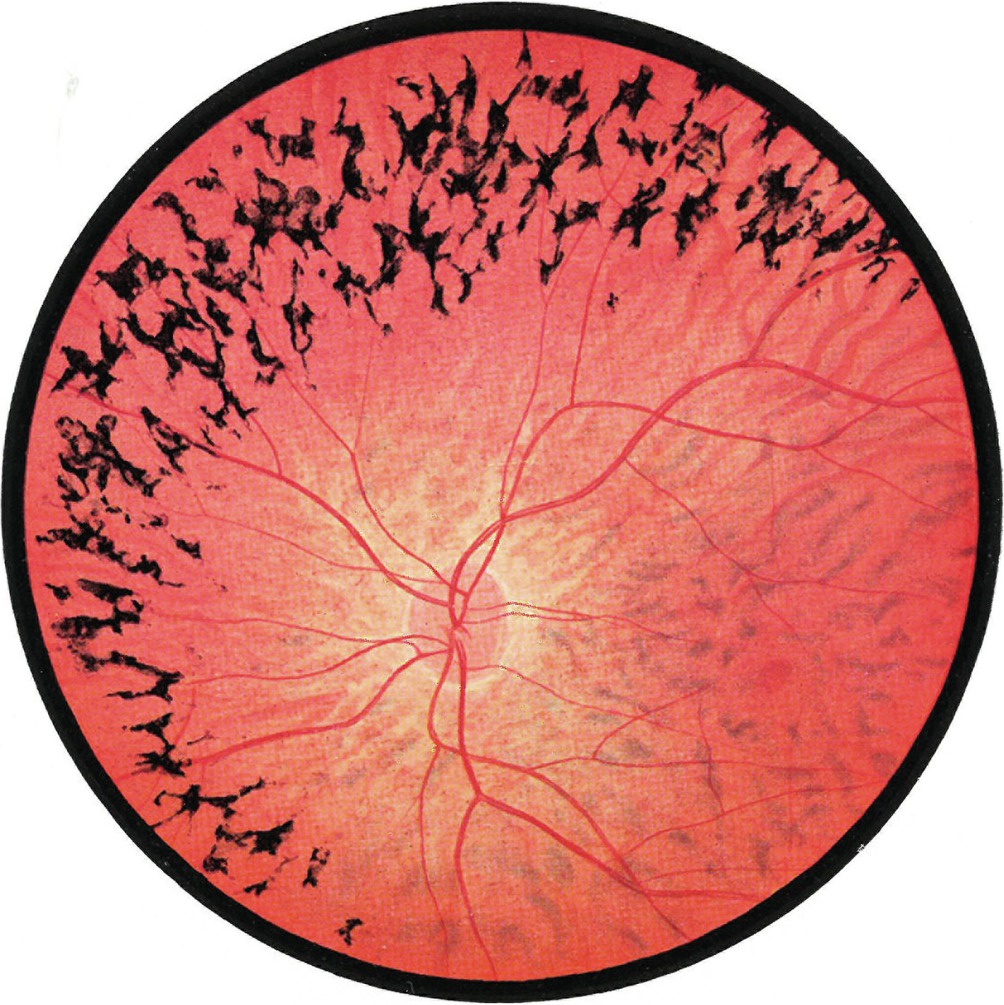
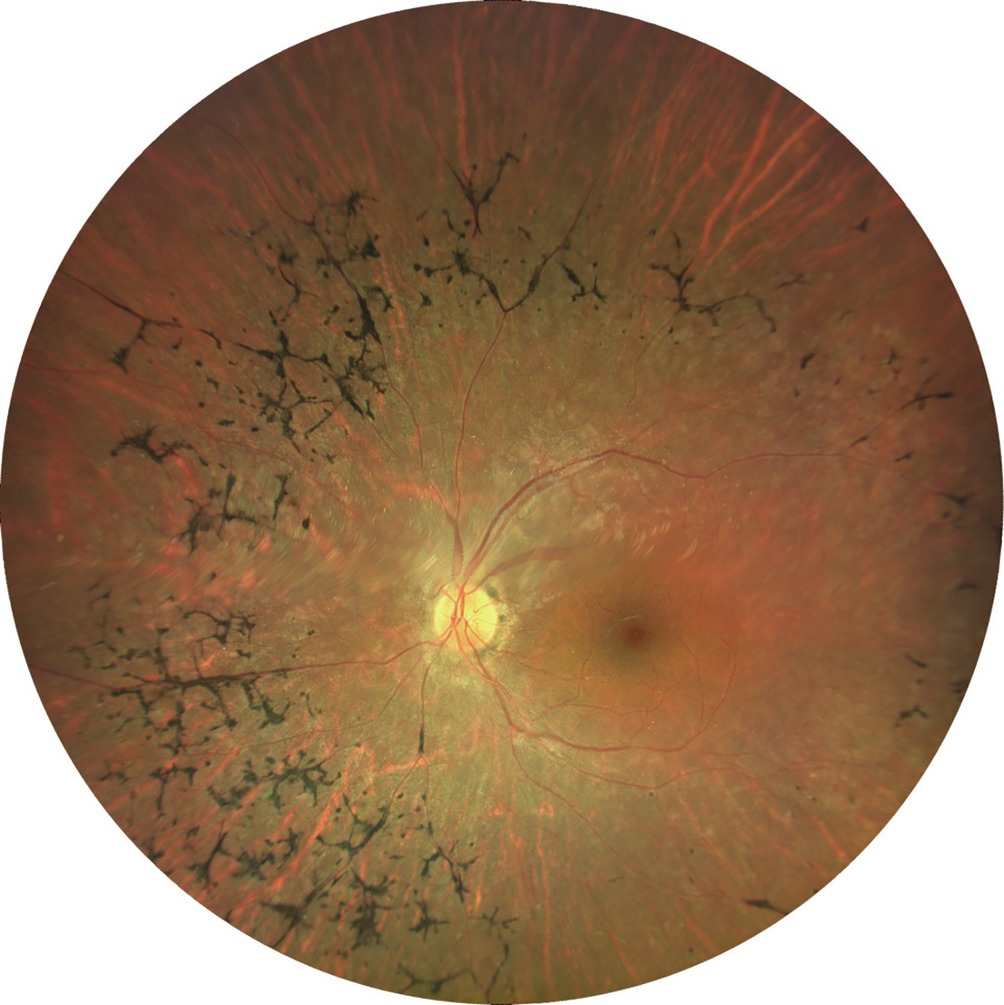
Alignement d'une illustration dessinée du rapport de 1963 et d'une photographie grand angle de 2026 de patients présentant des fonds d'œil similaires.
a. Reproduction de la figure 1, planche 1 du tome 1 du Rapport de la Société Française d'Ophtalmologie (SFO) 1963 d'Albert Franceschetti, Jules François et Jean Babel, Les hérédo-dégénérescences chorio-rétiniennes, Paris, Masson. La figure est intitulée «Rétinopathie pigmentaire en lumière ordinaire». b. Photographie grand angle (Clarus® Zeiss) du fond d'œil d'un patient de 35 ans porteur d'une variation pathogène dans le gène PRPF8 responsable d'une forme autosomique dominante de rétinopathie pigmentaire (CRMR CARGO, Strasbourg, 2026).